Auger Parameter
by M. Biesinger (UWO)
Following core ionization by photoelectron emission an outer shell electron can fill the created vacancy and the energy released can result in the emission of an Auger electron. A schematic of the Auger emission process for nickel metal is presented in Figure 1. The energy of an emitted Auger electron will be equal to the emitted photoelectron binding energy (Eb(C1)) minus the binding energy of electron that fills the vacancy in the core (Eb(C2)), minus the binding energy (in the presence of the core hole) of the level from where the Auger electron is emitted (Eb(C3)):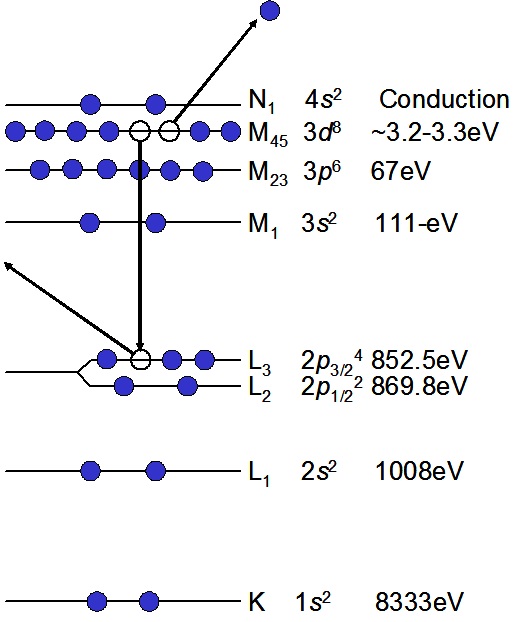
Ek ≈ Eb(C1) – Eb(C2) – Eb(C3) (1)
Figure 1. Schematic of an LMM Auger electron emission from a nickel atom.
Auger spectra have unique peak shapes and positions and are useful for both elemental identification and chemical state analyses. A calculated value from both photoelectron and Auger peak positions is the Auger parameter (α). This parameter is particularly useful for chemical state analysis and can be used without interference of surface charging. Originally defined by Wagner[1,2], the Auger parameter is calculated as follows:
α = Ek(C1C2C3) – Ek(C) (2)
where Ek(C1C2C3) is the kinetic energy of the Auger transition involving electrons from C1, C2 and C3 core levels and Ek(C) is the kinetic energy of the photoelectron from core level C. This form of the equation allowed for negative values of α. The Auger parameter was modified by Gaarenstroom and Winograd[3] by addition of the photon energy to α. This modified Auger parameter (α’) is independent of the X-ray energy used and is calculated as follows:
α’ = Ek(C1C2C3) + Eb(C) (3)
where Eb(C) is the binding energy of the core level C. Since any surface charging shifts will be of the same magnitude, but of opposite direction in each of these two components, they will be automatically cancelled out in α’.
The graphical display (scatter plot) of the most intense photoelectron line binding energies (abscissa, oriented in the negative direction) versus the kinetic energy position of the sharpest core-core-core Auger line (ordinate) is known as a Wagner plot, chemical state plot or chemical state diagram. Positions of compounds on these plots indicate both relaxation energy and initial state effects[4,5]. Hence, the modified Auger parameter can be used in addition to the binding energy envelope to give additional insight into the shift in electronic state between transition metal compounds.
There are numerous examples of the use of Wagner plots and Auger parameter in the literature including the study of silicon/silicate materials[6,7] and TiO2 on different supporting surfaces[8]. The NIST database[9] contains a large collection of Auger parameter values as does the Handbook of X-ray Photoelectron Spectroscopy[10].
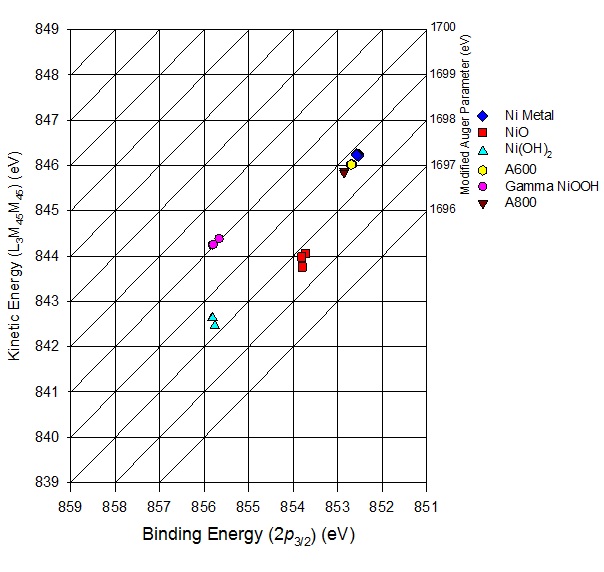
Figure 2. Ni 2p3/2 – Ni LMM Wagner plot for Ni metal, Ni alloys, NiO, Ni(OH)2 and NiOOH [From 11].
In XPS spectra, measured core level binding energies, Eb, involve both the ground state and the final state relaxation energies. The response of spectator electrons to the creation of a core hole and the Auger deexcitation process causes lowering of the measured binding energy as compared to the initial state (i.e. chemical shift) binding energy and this final state relaxation energy R can vary with chemical environment. Hence, there is a need to distinguish between initial and final state contributions to the measured binding energies. It is therefore important that final state effects are correctly described if binding energy shifts are to yield useful and reliable chemical information as to the electronic structure of transition metals and their compounds. Experimentally, relaxation energy shifts are often estimated by measuring the Auger parameter shift defined by:
Δα’ = ΔEb + ΔEk (4)
It is usually assumed, following the derivation by Morretti[4,5], that the relaxation energy for the doubly core-ionized state created by the Auger process, equals 2R, leading to:
Δα’ ≈ 2ΔR (5)
In the simplest approximation used by Wagner[12] and others[13,14] the shift in core level binding energy ΔEb and in Auger transition kinetic energy ΔEk are then:
ΔEb = − Δε – ΔR (6)
ΔEk = Δε + 3ΔR (7)
In this convention, positive values of Δε, initial state contributions, and ΔR, final state contributions, result in a shift to lower binding energy. Initial state effects, Δε, are generally understood to represent the “chemical shift” as a result of ground state electronic structure and are a function of the valence structure of the core atom, which is in turn is a function of bonding to neighboring atomic valence states. These shifts are related to the electronic states (e.g. band structures, bond directionality) and structural parameters (e.g. atomic positions, Madelung constants) of the bonded atoms.
References:
[1] C.D. Wagner, Electron Spectroscopy, in: D.A. Shirley (Ed.), Proceedings of an International Conference held at Asilomar, Pacific Grove, California, USA, 7-10 September, 1971, North-Holland, Amsterdam, 1972, p. 861
[2] C.D. Wagner, Anal. Chem. 44 (1972) 967.
[3] S.W. Gaarenstroom, N. Winograd, J. Chem. Phys. 67 (1977) 3500.
[4] G. Moretti, The Auger Parameter, in: D. Briggs, J.T. Grant (Eds.), Surface Analysis by Auger and X-ray Photoelectron Spectroscopy, IM Publications, Chichester, UK, 2003, pp. 501-530.
[5] G. Moretti, J. Electron Spectrosc. Relat. Phenom. 95 (1998) 95.
[6] P.S. Arora, R.St.C. Smart, Surf. Interface Anal. 24 (1996) 539.
[7] M. Stevenson, P.S. Arora, R.St.C. Smart, Surf. Interface Anal. 26 (1998) 1027.
[8] J.A. Mejías, V.M. Jiménez, G. Lassaletta, A. Fernández, J.P. Espinós, A.R. Gonzálex-Elipe, J. Phys. Chem. 100 (1996) 16255.
[9] C.D. Wagner, A.V. Naumkin, A. Kraut-Vass, J.W. Allison, C.J. Powell, J.R. Jr. Rumble, NIST Standard Reference Database 20, Version 3.4 (Web Version) (http:/srdata.nist.gov/xps/) 2003.
[10] J.F. Moulder, W.F. Stickle, P.E. Sobol, K.D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corp, Eden Prairie, MN, 1992.
[11] M.C. Biesinger, L.W.M. Lau, A.R. Gerson, R.St.C. Smart, Physical Chemistry Chemical Physics, 14 (2012) 2434.
[12] C.D. Wagner, J.A. Taylor, J. Electron Spectrosc. Relat. Phenom. 28 (1982) 211.
[13] J.S. Pan, J.G. Tao, C.H.A. Huan, Z. Zhang, J.W. Chai, S.J. Wang, Appl. Surf. Sci. 256 (2010) 4850.
[14] J.G. Tao, J.S. Pan, C.H.A. Huan, Z. Zhang, J.W. Chai, S.J. Wang, Surf. Sci. 206 (2008) 2769.