Multiplet Splitting
Discussion on Multiplet Splitting from chapter by Charles Fadley
C. S. Fadley, Electron Spectroscopy – Theory, Techniques and Applications, Vol 2, p1-156, 1978 published by Academic Press
MULTIPLET SPLITTING (split final state) is due to Interaction of Unpaired Electrons in Valence Levels and Unpaired Core Electron
Multiplet Splitting occurs in core level XPS whenever there is one (or more) unpaired electron(s) in the valence levels. Multiplet splitting occurs due to the exchange interaction between the unpaired valence electrons and the unpaired electron left in the core level (after photoionization). This interaction produces “split final states”.

Multiplet splittings arise from the various possible non-degenerate total electronic states that can occur in the final hole states of open-shell systems, whether they be atoms, molecules, or solids with highly localized unfilled valence levels.
The way in which multiple final states can be produced has already been briefly introduced in Section III.A, and for most systems it is adequate to consider a total spatial symmetry designation (e.g. L=O, l, 2, … in atoms), a total spin designation (e.g. S=O, I, 2, … in atoms or molecules), and perhaps also the perturbation of these via the relativistic spin-orbit interaction. The simplest interpretation of atomic multiplet splittings is thus in terms of various L, S terms.
Such effects can occur in any system in which the outer subshell or subshells are only partially occupied. The partial occupation provides certain extra degrees of freedom in forming total final states relative to the closed-shell case via coupling with the unfilled shell left behind by photoelectron emission. Multiplet effects can occur for both core and valence emission, as long as the valence subshell(s) are not totally occupied initially. Multiplet splittings also possess the important feature of being describable in first order in terms of a single set of ground-state Hartree-Fock one-electron orbitals. Thus, electron-electron correlation effects beyond the ground-state Hartree~Fock approximation are not essential for predicting that multiplet effects will exist, although, as will be shown, the inclusion of correlation effects is absolutely essential for quantitatively describing these phenomena in certain instances.
Multiplet effects involving core-level holes are very commonly encountered in interpretations of the fi ne structure an•s m• g m• x-ray em1· ss1· on spec tra243-245 and Auger electron spectra, 246-248 However, it is more recently that such effects were first recognized and studied in detail in connection with core x-ray photoelectron spectra of paramagnetic free molecules4• 249 and transition-metal compounds.as, 250 Subsequently, numerous studies have been carried out, including applications to systems containing both transition metal atoms, 1s1. 2so-257 and rare-earth atoms,156• 258-261 and a few comprehensive reviews have appeared.262-265 Primary emphasis here will be on the elucidation of a few examples to illustrate the types of effects noted and their modes of interpretation.
As an introductory example of one type of multiplet splitting found in XPS studies,. 2so consider first the ground-state Hartree-Fock description of photoemission from the 3s level of a Mn2+ free ion, as shown on the left hand side of Fig. 30. The ground state of this ion can be described in L, S (Russell-Saunders) coupling as 3d5 6S (that is, S= ½, L=O). In this state, the five 3d spins are coupled parallel. Upon ejecting a 3s electron, however, two final states may result: 3s3d5 5S (S=2, L=O) or 3s3d5 7S (S=3, L=O). The basic difference between these two is that in the 5S state, the spin of the remaining 3s electron is coupled anti-parallel to those of the five 3d electrons, whereas in the 7 S state the 3s and 3d spins are coupled parallel. Because the
exchange interaction acts only between electrons with parallel spins, the 7S energy will be lowered relative to the 5S energy because of the favorable effects of 3s-3d exchange. The magnitude of this energy separation will be proportional to the 3s-3d exchange integral Kas, aa, and will be given by
where e is the electronic charge, r < and r> are chosen to be the smaller and larger of r1 and r2 in performing the integrations, and Pas(r)/r and Paa(r)/r are the radial wave functions for 3s and 3d electrons. The factor 1/5 results from angular integrations involved in computing Kas, aa, A Hartree-Fock calculation of the energy splitting in Eq. (138) for Mn3+ gives a value of Mf(3s3d5)~ 13 eV.86, 250 As this predicted splitting is considerably larger than typical XPS linewidths, it is not surprising that rather large 3s binding energy splittings have in fact been observed in solid compounds containing Mn2+, and such splittings are clearly evident in the 3s regions of the first data of this type obtained by Fadley et a/.,86 , 250 as shown in Fig. 31. Roughly the left half of each of these spectra represents 3s emission, and the splittings observed in MnF2 and MnO are approximately one-half of those predicted from Eq. (138). The primary reason for this large discrepancy in magnitude appears to be correlation effects due to the highly overlapping character of the 3s and 3d orbitals, as discussed in more detail below.
In considering further such core binding energy splittings in non-relativistic atoms, it is worthwhile to present a more general discussion of the photoemission process, including the relevant selection rules.8• 262 • 263 If the photoelectron is ejected from a filled nl subshell containing q electrons, and an unfilled n’ I’ valence subshell containing p electrons is present, the overall
photoemission process can be written as:
Here, L and S denote the total orbital and spin angular momenta of the initial N-electron state and V and Sf represent the same quantities for the final ionic state with (N-1) electrons. As (n/)P is a filled subshell, its total orbital and spin angular momenta must both be zero and therefore L and S correspond to the orbital and spin momenta of the valence subshell (n’l’)P. In the final state, LI and Sf represent momenta resulting from the coupling of (n/)q-l (or, equivalently, a single core-electron hole) with (n’/’)P. The transition probability per unit time for photoelectron excitation is proportional to the square of a dipole matrix element between the initial and final state wave functions (see Section III.D.1 for a detailed discussion). In a nearly one-electron model of photoemission, this matrix element can be simplified to the sudden approximation forms given in Eqs (68) and (74). The selection rule on one-electron angular momentum is /1/ = // – / = ± 1, as stated previously. Conservation of total spin and total orbital angular momenta requires that and
Also, the overlap factors in Eqs (68) and (74) yield an additional monopole selection rule on the passive electrons, as introduced in Section III.D.1. This rule implies that the coupling of the unfilled valence subshell (n’l’)P in the final state must be the same as that in the initial state: that is to total spin and orbital angular momenta of Land S. Finally, any coupling scheme for (nl)q-l or (n’l’)P must of course be consistent with the Pauli exclusion principle. Since (n/)q-l is assumed to represent a single hole in an otherwise filled subshell, it must therefore couple to a total spin of½ and a total orbital angular momentum of l. Within this model, it has been shown by Cox and Orchard155 that the total intensity of a given final state specified by V, Sf will be proportional to its total degeneracy, as well as to the one-electron matrix element squared. Thus, in Russell-Saunders coupling
For the special case of atomic s-electron binding energy splittings, the relevant selection rules are thus:
and the total intensity of a given peak is predicted to be proportional to the spin degeneracy of the final state:
Thus, only two final states are possible corresponding to SI= S ± ½, and the relative intensities of these will be given by the ratio of their multiplicities,
or
The energy separation of these two states can further be calculated from simple atomic multiplet theory and is given by a result often referred to as the Van Vleck Theorem:11s
Here Kns, n’z’ is the ns-n’l’ exchange integral and can be calculated from where the same notation as that in Eq. (138) has been used. Equations (146)-(150) indicate that such s-electron binding energy splittings should yield a doublet with a more intense component at. lower binding energy (corresponding to an exchange-favored final state of Sf=S+½) and a component separation that is directly associated with both the initial state spin and the spatial distributions of the core and valence electrons as reflected in the exchange integral. Thus, the potential for extracting certain types of useful and unique information from such splittings exists. That Eq. (148) provides a good description of the systematics of such s-Ievel multiplet splittings has been nicely demonstrated in studies of the 4s and 5s splittings in rare-earth metals and compounds with varying outer. 4f subshell occupation numbers and spins S, 258, 259 as summarized in Fig. 32.
Experimental (points) and theoretical (lines) 4s and 5s binding energy splittings m various rare-earth ions. The 8Evv values are calculated using Van Vleck’s Theorem [Eq. (148)]. Experiment and theory are in excellent agreement for 5s but the theoretical splittings must be reduced by a factor of 0·55 to agree with the 4s data because of correlation
effects. (From McFeely et al., ref. 259.)The solid lines connect calculated values based upon Eq. (148) and are in excellent agreement with experiment for the 5s splittings, whereas for the 4s
splittings, this simple theory must be reduced by a factor of ~0·55 to agree with experiment. These results also suggest that the 4s discrepancy may be due to the same type of correlation correction involved in Mn3s, as the 4s-4f spatial overlap is high, increasing correlation, whereas the 5s-4f overlap is much lower, decreasing it. Configuration interaction calculations on Mn3+ by Bagus et al.252 first provided a more quantitative understanding of such correlation corrections to intra-shell s-level splittings such as 3s-3d and 4s-4f. They pointed out that, in a Cl description of the true Mn3+ final states corresponding to 3s emission, several configurations would be of special importance in addition to the usual one-electron-transition final configuration as shown in the left half of Fig. 30.
(In writing such configurations below, numbers in parentheses will denote the L, S coupling of the subshell to the left.) The 7S final state is found to be composed almost completely of 3s1(2S)3p6(1S)3d5(6S), the one electron configuration, and so is not much perturbed by CL Another way of saying this is that there is already strong exchange correlation in 7S, so that the addition of CI is not so significant. The 5S final state is by contrast expected to have significant contributions from not only the one-electron configuration <l>1(1>S) = 3sl(2S)3p6(1S)3d5(6S), but also from configurations in which it formally appears that one 3p electron has been transferred down to a 3s orbital while another 3p electron has been transferred up to a 3d orbital:
(The notations 3d6(3P1) and 3d6( 3P2) stand for two independent ways in which 3d6 can couple to 3P.)
Thus, there will be at least· a fourfold manifold of 5S states, and the lowest-energy member is expected to be lowered significantly (that is, to move toward 7S). In fact, the 5S state nearest 7S is found to be only 4·71 eV away, in much better agreement with the experimental splitting for MnF2 of 6·5 eV than the estimate of ~ 13 eV obtained from Eq. (138). Such intrashell s-level multiplet splittings can thus only be predicted accurately when correlation is allowed for, whereas inter-shell s-level splittings are, by contrast, well predicted by Eq. (148). A further significant effect predicted by these Cl calculations for the Mn3+ 5S states is the existence of additional experimental fine structure. Specifically, there are four 5S states at £1, £2, Ea, and £4, that can be written to a good approximation asAs the initial state is rather well described by a single configuration 3s2(1S)3p6(1S)3d5( 6S) possessing the d-electron coupling of <J.)1, the sudden approximation result of Eq. (84) can immediately be used to show that the four 5S intensities will be given by
with the total intensity Ii+ h +Ia+ /4 still being proportional to the spin degeneracy of 5. Evaluating the energies and relative intensities in this way yields a prediction of a total of only three observable 5S peaks (one is too weak to be seen easily) and one observable 7 S peak in the Mn2+ spectrum. Weak structures in . good agreement with these predictions have, in fact, been observed by Kowalczyk et al.,253 and their experimental results are shown in Fig. 33. These Cl calculations also explain a peak intensity discrepancy noted relative to simple multiplet theory: namely that the intensity ratio 5S(l)/7S in Fig. 31 or Fig. 33 is significantly below the 5/7 predicted by
Higher resolution Mn3s spectrum from MnF2 obtained with monochromatized Al K radiation (cf. Fig. 31). The peaks 5S(2) and 5S(3) arise from final-state configuration interaction (correlation effects) according to Eq. (151). (From Kowalczyk et al., ref. 253.) Eq. (146). It is thus clear, that, although a first-order description of multiplet effects is possible within a non-correlated Hartree-Fock approach, a detailed description of the numbers, positions, and relative intensities of peaks , may require including correlation effects, especially where intra-shell interactions dominate. The first observations of s-electron core binding energy splittings analogous to those described by Eqs (146)-(150) were in gaseous, paramagneticmolecules.4, 249 Hedman et a/.249 found splittings as large as l ·5 eV in the ls photoelectron spectra of the molecules NO and 02. These results are shown in Fig. 34 along with an unsplit ls spectrum from the diamagnetic molecule N2. In each case, it can be shown that the observed energy splitting should be proportional to an exchange integral between the unfilled valence molecular orbital and the ls orbital of N or 0,4 in analogy with Eq. (148).
Theoretical estimates of these splittings from molecular orbital calculations give values in good agreement with experiment,4, 107 as expected for such inter-shell interactions in which correlation effects are much decreased. The observed intensity ratios of the peaks are furthermore very close to the ratios of the final-state degeneracies, also in agreement with simple theory.
The analysis of binding energy splittings in emission from non-score levels is not as straightforward as for s-level emission, primarily due to the fact that the core-electron hole represented by (nl)q-l (which now has associated with it a spin of½ and a non-zero orbital angular momentum of/) can couple in various ways with the valence subshell (n’l’)P (which can have various spins S” and orbital angular momenta L”, including the initial values Sand L) to form a final state with a given total spin St and total orbital angular momentum U. Thus, the number · of allowed final states increases and their energy separations will in general be determined by both Coulomb and exchange integrals through different coupling schemes. Additional complexities arising for non-s levels are caused by spin-orbit coupling and crystal-field splittings.
The simplest procedure for calculating such non-s energy separations is again to use non-relativistic atomic multiplet theory.86, 2so, 262, 263 As an illustrative example, consider 3p electron emission from Mn2+, as indicated in the right-hand portion of Fig. 30. For this case, (nl)q-1=3p5, (n’l’)P=3d5 and the initial state, as before, is 6S (S=½, L=0). The previously stated selection rules imply that the allowed final states correspond to 7 P(S = 3 L=l) and 5P(S=2,L= l). Although a 5S(S=2,L=0) final state would be, consistent with selection rule (141), it requires changing the coupling of 3d5 from its initial 6S and so is not allowed. There is only one way for 3p5 to couple with 3d5 to form a 7 P state, that being with 3p5 (always coupled to total spin and total orbital angular momentum=/= l) coupled with 3d5 in its initial state coupling of 6S (S=½, L=0). However, there are three ways to form the allowed 5P final state by coupling
Thus, four distinct final states are possible for 3p emission from Mn2+, one 7 P and three 5 P. As there are off-diagonal matrix elements of the Hamiltonian between the various 5P coupling schemes,118 they do not individually represent eigenfunctions. The eigenfunctions describing the 5 P final states will thus be linear combinations of the three schemes:
where each 5 P configuration has been labelled by the 3d5 coupling involved and the C,/s are the usual expansion coefficients. The energy eigenvalues corresponding to these eigenfunctions will give the separations between the 5P states. Such eigenfunctions and eigenvalues can most easily be determined by diagonalizing the 3 x 3 Hamiltonian matrix for the 5 P states, where each matrix element is expressed as some linear combination of Jad,, 3d,, Kaa, aa, Jap,3d, and Kap, aa.33, 118 If Coulomb and exchange integrals from a Hartree-Fock calculation on Mn2+ are used, such matrix diagonalization calculations yield the relative separations indicated on the right-hand side of Fig. 3086, 250 Once again, the sudden approximation result of Eq. (84) indicates that,
because the initial state is rather purely 3d5(6S), only those components of the 5P states represented by Cn(J)(6S) are accessible. Thus, the individual intensities of ‘¥1, ‘¥2, and ‘Ya can be computed from jCul2, jC21 l2, and I Cai j 2, respectively. In determining the total intensity ratios for the 5P and 7P states, Eq. (142) can be used to give: The relative peak heights in Fig. 30 have been calculated in this way, and the experimental 3s(l )-3p(1) separation and relative intensity for MnF2 were used to empirically fix the scales between the 3s and 3p regions. The separations and relative intensities of the peaks observed are found to be at least semi-quantitatively predicted by this simple, atomic L, S coupling model, 86, 250 and these results have been confirmed in more detail by later experimenta1255 and theoretical256 studies.
The remaining discrepancies between theory and experiment for this 3p case could be caused by a combination of effects due
to correlation, spin-orbit coupling, and crystal-field splitting, although calculations by Gupta and Sen256 indicate that the latter two are probably not so significant: Ekstig et aL245 have carried out matrix diagonalization calculations like those described here but for more complex sets of final 3p-hole states in 3d transition metal atoms in an attempt to interpret soft x-ray emission spectra from solids. The theoretical aspects of calculating such non-s splittings have also recently been reviewed by Freeman et al. 263 Deeper non-s core levels in 3d atoms should also exhibit similar splittings, although the magnitudes will be reduced because of the decreased interaction strengths between the core and 3d orbitals. For example, Fadley and Shirley86 first noted that the Mn2p levels in MnF2 are broadened by ~ l ·5 eV relative to those in low-spin (filled subshell) compounds, and suggested multiplet splittings as the origin of this broadening. Subsequent measurements at higher resolution by Kowalczyk et al., 255 coupled with theoretical calculations by Gupta and Sen,257 have confirmed this suggestion, and also verified the existence of peak asymmetries and anomalous 2pr2Pt separations.
For this 2p case, both multiplet effects and spin-orbit coupling are of similar magnitude, and were included in calculations that successfully predicted the observed spectra.257 Analogous non-s core-level splittings have also been studied in systems with partially-filled f subshells, 86, 260, 266 and the anomalous shape and decreased spin-orbit splitting in the Eu4d spectrum of Fig. 6 is, ,in fact, attributable to such effects. 86 Although only multiplet effects on core-level binding energies have been considered up to this point, such phenomena can play a considerable role in determining the fine structure observed in valence spectra (as has been apparent for some time in UPS studies of free molecules97). In particular, XPS valence spectra obtained from solids containing highly localized d levels or /levels are expected to be influenced by such multiplet effects,82, 156, 157, 1 61, 266, 267 with the relative intensities of various allowed final states being determined by fractional parentage coefficients, as described in Section 111.D.2 and elsewhere.156, 157, 262 Heden et a/.267 first observed such effects in valence spectra of 4f metals. As an example of the occurrence and use of such splittings in studies of rare-earth compounds, the XPS results of Campagna et al. 261 and Chazalviel et al. 266 show strong multiplet splittings in the valence spectra of Sm-chalcogenides and a mixture of two markedly different multiplet structures in certain Sm compounds that are thought to exhibit valence fluctuations between Sm+2 4f7 and Sm+s 4f6,
Some of these results for SmB6266 are presented in Fig. 35, in which the L, S multiplets expected for both Sm+2 and Sm+3 are labelled. Theoretical intensities have been calculated using fractional parentage coefficients, 156 and the agreement between the theoretically simulated spectrum and experiment is excellent. Baer126~ has also presented very high-resolution XPS spectra for various 4f metals that further confirm the existence of these atomic-like multiplet effects. In analogous multiplet effects in valence d orbitals, the inclusion of crystal-field effects is also expected to be important, as has been emphasized in a recent discussion by Bagus et a/.157 In comparison to chemical shifts of core-electron binding energies, multiplet splittings of core- or valence-energies thus represent higher-order effects yielding a different type of information. In their simplest interpretation, chemical shift measurements detect a change in the spatially-averaged potential experienced by an electron, whereas analyses of multiplet effects have the capability of determining the valence electron configuration or the detailed strengths of various higher-order electronic interactions. The two types of measurements are thus complementary. Numerous applications of multiplet splittings measurements are thus possible in the study of the transition series metals, the rare earths, the transuranium elements, and open-shell systems in general.
Multiplet Splitting
by M. Biesinger (UWO)
Multiplet splitting arises when an atom contains unpaired electrons (e.g. Cr(III), 3p63d3). When a core electron vacancy is created by photoionization, there can be coupling between the unpaired electron in the core with the unpaired electrons in the outer shell. This can create a number of final states, which will be seen in the photoelectron spectrum as a multi-peak envelope[1]. Figure 1 shows the multiplet structure associated with the Cr 2p3/2 peak for a vacuum fractured Cr2O3 specimen.
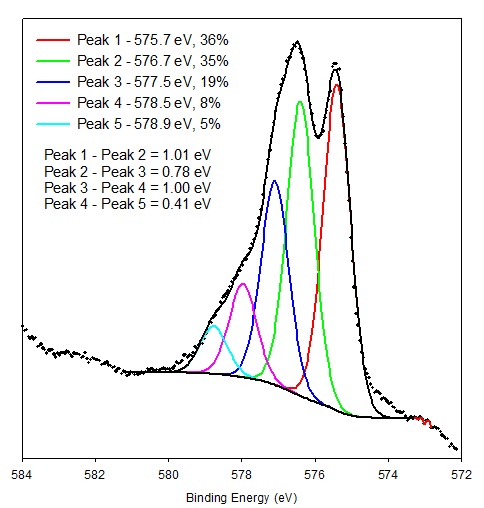
Figure 1. Multiplet structure associated with the Cr 2p3/2 peak for a vacuum fractured Cr2O3 specimen.
The early Hartree-Fock calculation of the multiplet structure of core p-valence levels of free ion state first row transition metals by Gupta and Sen[2] graphically shows their multiplet structures (Figure 2). These calculations are an excellent starting point for the examination of multiplet structure observed for transition metal compounds. However, they apply to free ion states only and, in transition metals and their compounds, there may be ligand charge transfer effects that will change the spacing and intensity of the multiplet peaks present in their spectra. These relative changes can be utilized for transition metal compounds to differentiate those more closely approximating free ions from those in which charge transfer from the bonded neighbouring ions may have changed both the effective oxidation state and multiplet splitting of the core transition metal[3,4,5,6]. This change in local electronic structure has been used to explain the differences between the XPS spectra of nickel oxide and its oxy/hydroxides [3,7]. De Groot and Kotani’s text “Core Level Spectroscopy of Solids”[8] provides an excellent advanced analysis of multiplet effects and their use in the modeling of spectra for both XPS and XAS.
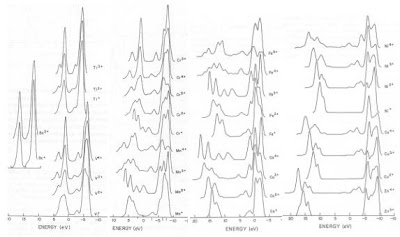
Figure 2. Calculated multiplet structure of 2p ionisation created in the free ions as labelled. The zero energy is arbitrary and the intensity normalization is the same for all spectra shown [From 2].
Table 1 summarizes the various first row transition metal species that show multiplet splitting in their XPS spectra. Sc, Ti, V, Cu and Zn species, where multiplet splitting is not present or, if present, is generally not well resolved or shows as peak broadening only[9]. Cr, Fe, Mn, Co and Ni species show significant multiplet spitting[3,4,5,6,7,10,11].
![]()
Table 1. First row transition metal species that show multiplet splitting in their XPS spectra. This is for high spin compounds. For low spin Fe(II) and low spin Ni(II) electrons are paired and no multiplet splitting is observed.
References:
[1] J.F. Moulder, W.F. Stickle, P.E. Sobol, K.D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corp, Eden Prairie, MN, 1992.
[2] R.P. Gupta, S.K. Sen, Phys. Rev. B 12 (1975) 15.
[3] A.P. Grosvenor, M.C. Biesinger, R.St.C. Smart, N.S. McIntrye, Surf. Sci. 600 (2006) 1771.
[4] N.S. McIntyre, D.G. Zetaruk, Anal. Chem. 49 (1977) 1521.
[5] M.C. Biesinger, C. Brown, J.R. Mycroft, R.D. Davidson, N.S. McIntyre, Surf. Interface Anal. 36 (2004) 1550.
[6] A.P. Grosvenor, B.A. Kobe, M.C. Biesinger, N.S. McIntyre, Surf. Interface Anal. 36 (2004) 1564.
[7] M.C. Biesinger, L.W.M. Lau, A.R. Gerson, R.St.C. Smart, Physical Chemistry Chemical Physics, 14 (2012) 2434.
[8] F. de Groot, A. Kotani, Core Level Spectroscopy of Solids, CRC Press, Boca Raton, 2008.
[9] M.C. Biesinger, L.W.M. Lau, A.R. Gerson, R.St.C. Smart, Appl. Surf. Sci. 257 (2010) 887.
[10] M.C. Biesinger, B.P. Payne, A.P. Grosvenor, L.W.M. Lau, A.R. Gerson, R.St.C. Smart, Appl. Surf. Sci. 257 (2011) 2717.
[11] M.C. Biesinger, B.P. Payne, L.W.M. Lau, A.R. Gerson, R.St.C. Smart, Surf. Interface Anal. 41 (2009) 324.